02 - GenBank format and annotation¶
GenBank format¶
When a new genome sequence gets published, it should be deposited in the global sequence archive under the International Nucleotide Sequence Database Collaboration (INSDC), via one of the three mirrors run by the NCBI in America, EMBL-EBI in Europe, and DDBJ in Japan.
We're going to be using the NCBI website here, and the NCBI default view here shows the whole genome as a webpage with clickable links based on the plain text NCBI GenBank format.
GenBank format is intended to be human readable (see this sample record), and is a widely used file format for annotated genomes. The related EMBL file format used in the European sequence database which mirrors the NCBI sequence database shares the same INSDC feature table design.
A sequence record in GenBank format has three main sections:
- header starting with the
LOCUSline - feature table listing any annotated features like genes
- the actual sequence (or how it is built up from other records), ending with a
//line.
With bacterial genomes, for each annotated gene you expect to see a pair of features - a gene immediately followed by a CDS entry for the coding sequence, using the same location co-ordindates. With eukaryotes in addition there is usually an mRNA entry, and the CDS location is more complicated in order to describe only the exons which make up the coding sequence.
Here we've picked a Pectobacterium atrosepticum (previously known as Erwinia carotovora) genome as an example, NC_004547.2. You can scroll down to see the genes, or search within the page once it has fully loaded.
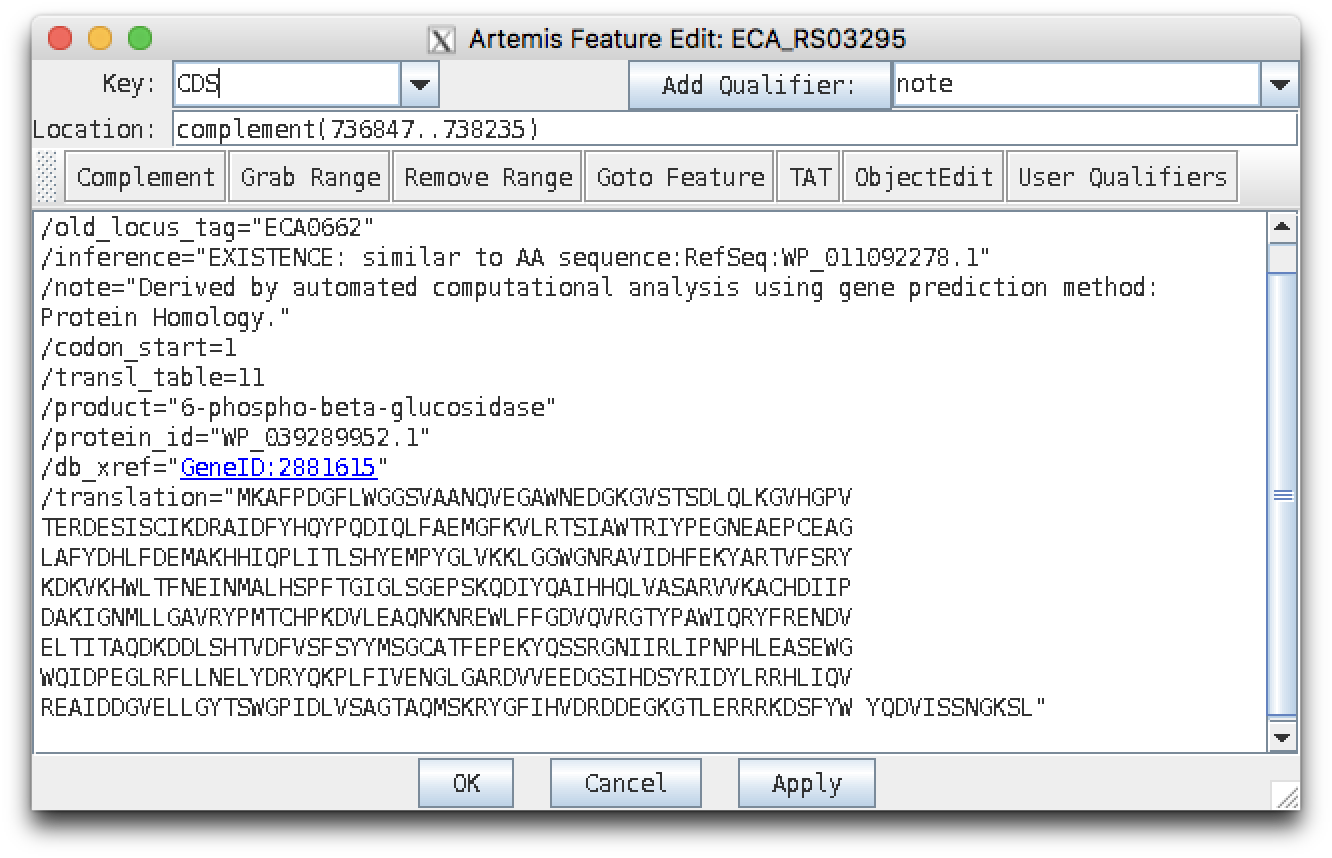
Supposing you were interested in the gene with original locus tag ECA0662 (which is the first entry in example file glycoside_hydrolases_nt.fasta), you should find it here:
gene complement(736847..738235)
/locus_tag="ECA_RS03295"
/old_locus_tag="ECA0662"
CDS complement(736847..738235)
/locus_tag="ECA_RS03295"
/old_locus_tag="ECA0662"
/inference="COORDINATES: similar to AA
sequence:RefSeq:WP_010282995.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="6-phospho-beta-glucosidase"
/protein_id="WP_039289952.1"
/translation="MKAFPDGFLWGGSVAANQVEGAWNEDGKGVSTSDLQLKGVHGPV
TERDESISCIKDRAIDFYHQYPQDIQLFAEMGFKVLRTSIAWTRIYPEGNEAEPCEAG
LAFYDHLFDEMAKHHIQPLITLSHYEMPYGLVKKLGGWGNRAVIDHFEKYARTVFSRY
KDKVKHWLTFNEINMALHSPFTGIGLSGEPSKQDIYQAIHHQLVASARVVKACHDIIP
DAKIGNMLLGAVRYPMTCHPKDVLEAQNKNREWLFFGDVQVRGTYPAWIQRYFRENDV
ELTITAQDKDDLSHTVDFVSFSYYMSGCATFEPEKYQSSRGNIIRLIPNPHLEASEWG
WQIDPEGLRFLLNELYDRYQKPLFIVENGLGARDVVEEDGSIHDSYRIDYLRRHLIQV
REAIDDGVELLGYTSWGPIDLVSAGTAQMSKRYGFIHVDRDDEGKGTLERRRKDSFYW
YQDVISSNGKSL"We want to learn more about the meaning of this entry, and the gene context. First, since we're going to work more with this GenBank file directly, we'll need to download it.
Downloading GenBank files¶
You could download the the GenBank file we want via the NCBI website, but it takes a lot of manual steps. Currently you would start with the "send" menu to the top right of screen, picking "Complete Record", destination "File", and format "GenBank (full)". Save the file as NC_004547.gbk.
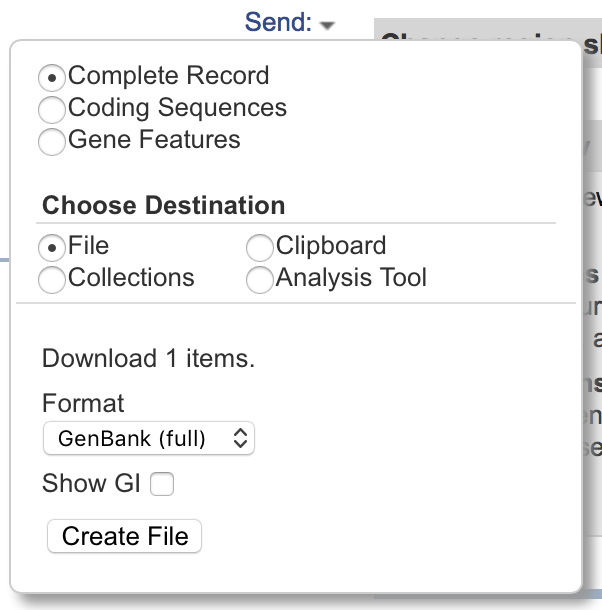
If you accidently use format "GenBank", then with these examples you'll get a much smaller file missing all the features and sequence data. Also, depending on your browser's settings, the file may be saved with a default name like sequence.gb.txt in your downloads folder, which means you'd have to move the file to the working folder and rename it.
Thankfully the NCBI provide a way to automate this, which they call the NCBI Entrez Programming Utilities. This can be used from many different programming languages, but we will use it from Python using the Biopython wrapper Bio.Entrez to help.
########################################################################
# For the purposes of this workshop, don't worry about how this works, #
# a key point here is if you run this you'll all get the same file. #
########################################################################
# Biopython's module to access the NCBI Entrez Programming Utilities
from Bio import Entrez
# The NCBI likes to know who is using their services in case of problems,
Entrez.email = "your.name.here@example.org"
accession = "NC_004547"
print("Fetching %s from NCBI..." % accession)
# Return type "gbwithparts" matches "GenBank (full)" on the website
fetch_handle = Entrez.efetch("nuccore", id=accession, rettype="gbwithparts", retmode="text")
# Open an output file, and write all the data from the NCBI to it
with open(accession + ".gbk", "w") as output_handle:
output_handle.write(fetch_handle.read())
print("Saved %s.gbk" % accession)
If you look at NC_004547.gbk in a text editor, or simply view it at the command line using less, it should look like the NCBI website's content - although there are some interesting differences...
$ cd ~/2018-03-06-ibioic/01-introduction
$ less NC_004547.gbk
...
Genomic context¶
You can look directly at the GenBank file see what genes or other features are annotated nearby, and deduce if these genes are part of an operon, or gene locus etc. However, this is much easier to see graphically.
Genomic context on the NCBI website¶
Until May 2017, the GenBank entry for this gene ECA0662 we searched for had a clickable database cross-reference link, GeneID:2881615 - but the NCBI have retired all the GI numbers (public records and databases are often subject to change, which is one reason why it's important to specify accession identifiers, and dates of access in publications). Anyway, for now that link still shows this gene on the NC_004547.2 genome visually:
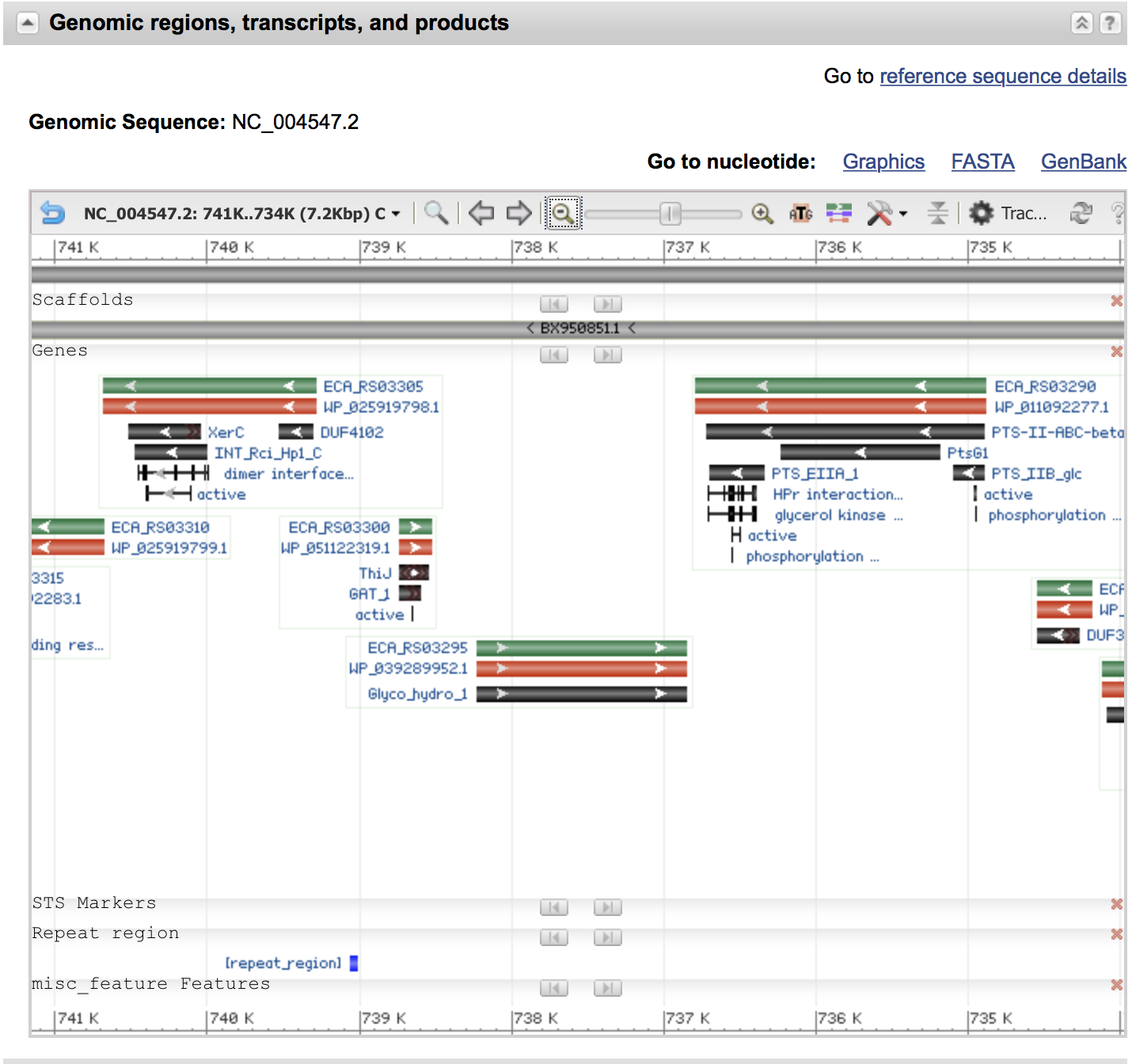
You can try zooming out etc to examine what else is nearby - is your gene of interest part of an operon? Are the repeat regions nearby or anything else which might be important like an integrated phage?
However, nice as that is, it is often useful to be able to vizualise the genomic context outwith the NCBI website. You might be working on a draft genome which has not been published yet, for example.
For this we will open the downloaded GenBank plain text files in the Sanger Institute's tool Artemis, which is able to view and edit annotation files.
Genomic context using Artemis¶
You should have previously download this GenBank file, in which case you can load it with Artemis via the command art as follows:
$ cd ~/2018-03-06-ibioic/01-introduction
$ ls *.gbk
NC_004547.gbk
$ art NC_004547.gbk &
Artemis is a graphical tool written in Java, and should pop up a separate window.
Windows users, you will instead probably have to launch the artemis.jar file, and then from the "File" menu, pick "Open..." and navigate to the NC_004547.gbk file.
After a couple of harmless warnings, you should get a window like this:
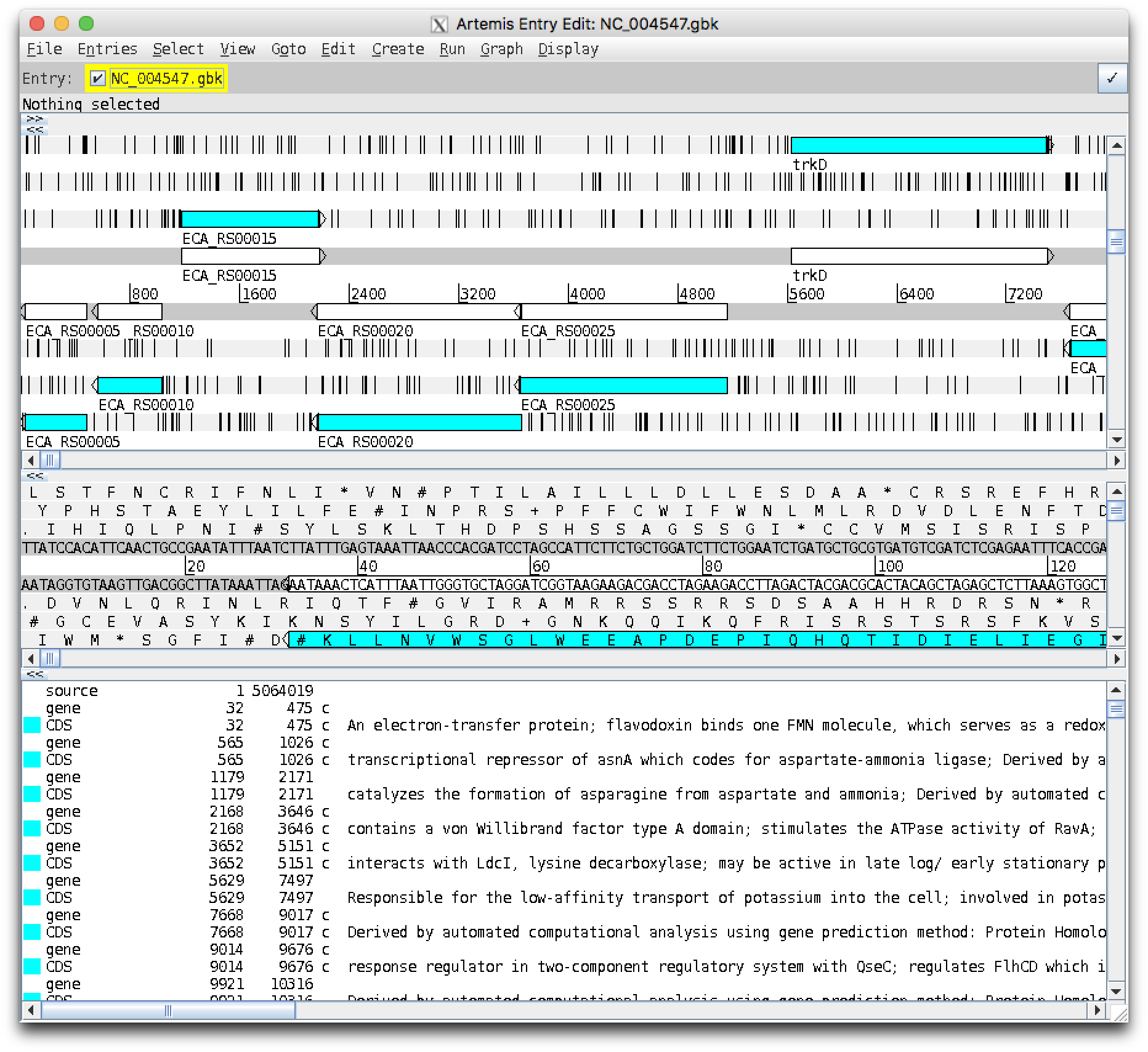
The main window should have three regions,
- Zoomed out view (showing genes as coloured bars)
- Zoomed in view (showing individual bases and translated reading frames)
- List of features
Using the menu entry "Goto", "Navigator..." (or by pressing Ctrl+g), which pops up an "Artemis Navigator" window, tell Artemis to goto feature with the qualifier value ECA0662 (the old locus ID), and you should see something like this:
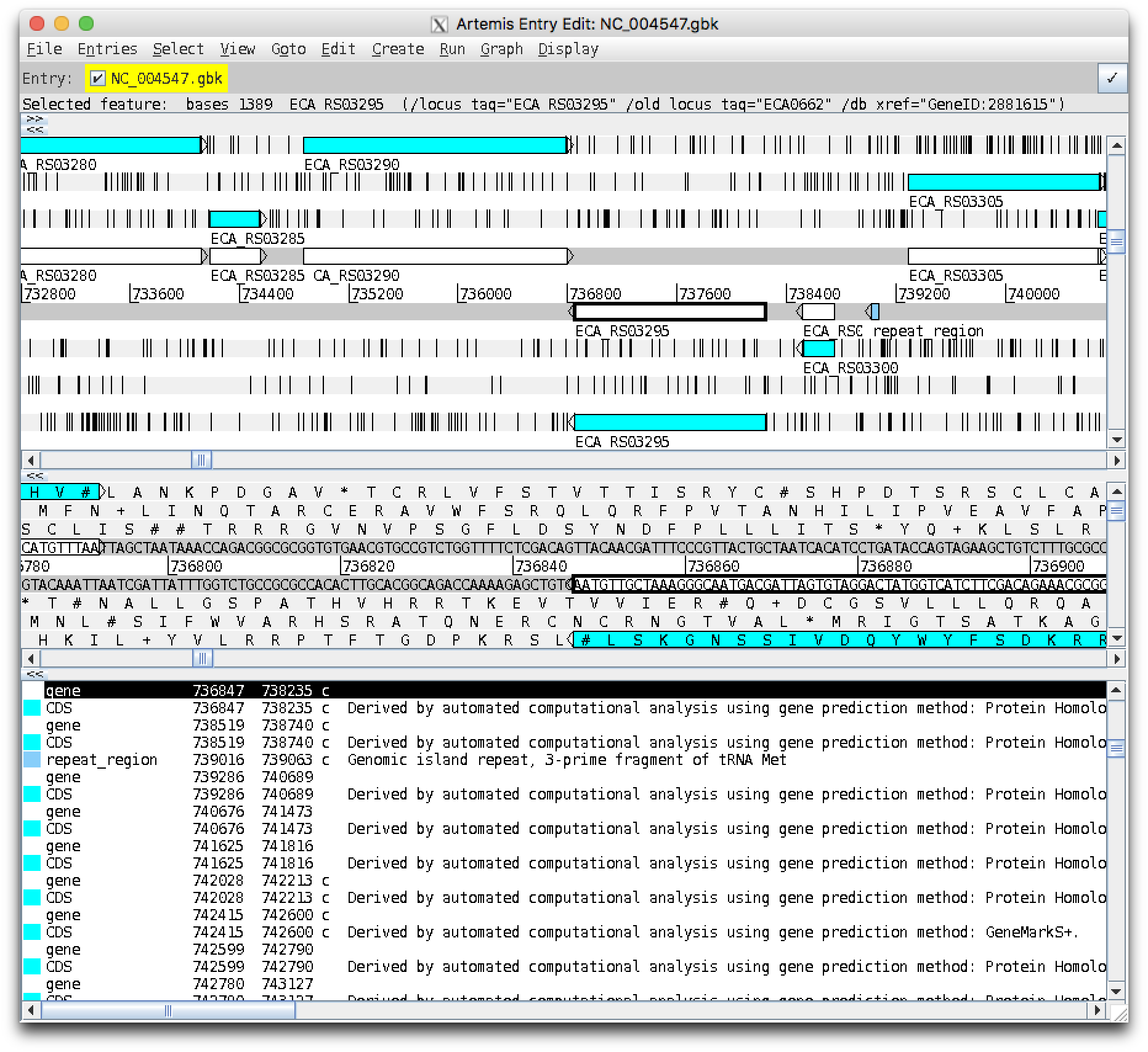
Notice a white gene feature has been outlined in black. Click "Goto" again, and the pale blue CDS feature should be highlighted:
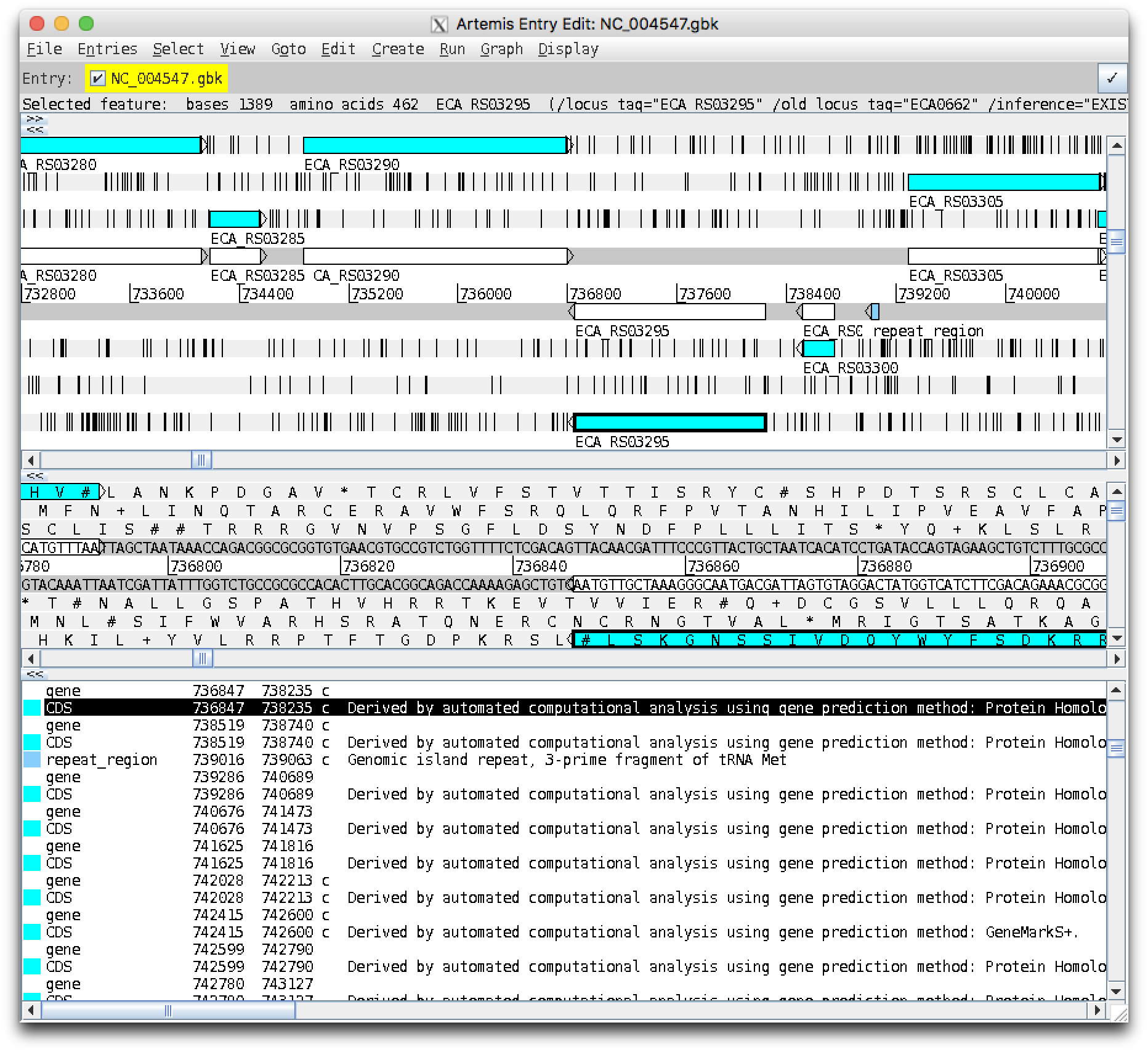
Clicking "Goto" a third time should say "text not found".
This gene is on the reverse complement strand - Artemis shows these features on the bottom half of the track, with the arrow pointing left. Looking to the left (downstream) there is a gene on the opposite forward strand. Looking to the right (upstream) there is a small gene on the same strand, then a small steel blue repeat region.
In the bottom pane Artemis shows any note in the annotation for each feature - this repeat region is described as a genomic island repeat, 3-prime fragment of tRNA Met.
If you double click on any feature, Artemis will jump to center it.
With a feature selected, pressing E brings up a window to edit the annotation of that feature. This is the quickest way to see things like the product or other information.
Here's the edit view for the ECA0662 CDS feature, which should be very familiar from looking at the GenBank file format entry: