
04 - Multiple Sequence Alignment¶
Table of Contents¶
Introduction¶
To recap, we've downloaded the Pectobacterium atrosepticum genome in GenBank format as NC_004547.gbk, and then extracted eight glycoside hydrolase genes from it as two FASTA files with their nucleotide and protein seqeunces giving files nucleotides.fasta and proteins.fasta.
When you have a set of genes you think form a gene family, a natural thing to do it compare the sequences to each other. A standard approach here is to build a multiple sequence alignment, and while there are many tools for this we will use muscle.
These tools will generally work even on a pair of sequences, but here there even more options and ways to visualise the similarities or differences in a pairwise alignment. You should probably start with a dotplot.
Running Muscle¶
Muscle is a popular tool for multiple sequence alignment of nucleotides or proteins. By default it expects the input sequences in FASTA format, and will produce the output alignment in FASTA format where minus signs - have been inserted into the sequences to indicate gaps the alignment introduces. Let's try this with our proteins:
$ cd ~/2018-03-06-ibioic/01-introduction/
$ muscle -in proteins.fasta -out proteins_aligned.fasta
MUSCLE v3.8.31 by Robert C. Edgar
http://www.drive5.com/muscle
This software is donated to the public domain.
Please cite: Edgar, R.C. Nucleic Acids Res 32(5), 1792-97.
proteins 8 seqs, max length 1473, avg length 1423
00:00:00 1 MB(0%) Iter 1 100.00% K-mer dist pass 1
00:00:00 1 MB(0%) Iter 1 100.00% K-mer dist pass 2
00:00:00 12 MB(0%) Iter 1 100.00% Align node
00:00:00 12 MB(0%) Iter 1 100.00% Root alignment
00:00:01 14 MB(0%) Iter 2 100.00% Refine tree
00:00:01 14 MB(0%) Iter 2 100.00% Root alignment
00:00:01 14 MB(0%) Iter 2 100.00% Root alignment
00:00:01 15 MB(0%) Iter 3 100.00% Refine biparts
00:00:02 15 MB(0%) Iter 4 100.00% Refine biparts
00:00:02 15 MB(0%) Iter 5 100.00% Refine biparts
You should now have a new file proteins_aligned.fasta which you can look at within the command line terminal using less:
$ less proteins_aligned.fasta
>ECA3646
ATGAGTAAGGTTTCACTGAC------TA---TTCCCCCAGATTTTATTCTGGGTGCGGCG
GCATCGGCATGGCAAACGGAAGGATGGAGCGGCAAGAAGGCAGGGCAGGAT--TCCTATT
TGGATCTC-------------------------------------TGGTACAAGCAG---
...
This isn't intended to be interpreted by eye - the best way to view this is with a graphical tool, and here we will use Jalview.
Running Jalview¶
In this workshop, we're going to use Jalview only as a multiple sequence aligment viewer, but it is capable of much more.
If it is not installed locally (or you are using Windows) try this Jalview Launch link, then use the application menu "File", "Input Alignment", "from File" to pick your proteins_aligned.fasta file.
However, if it is installed locally on Linux or macOS, the quickest and easiest way to open the file directly in Jalview is via the command line:
$ jalview -open proteins_aligned.fasta
...
This will print lots of debugging information in the terminal, but also pop up the main Jalview window with our protein alignment loaded:
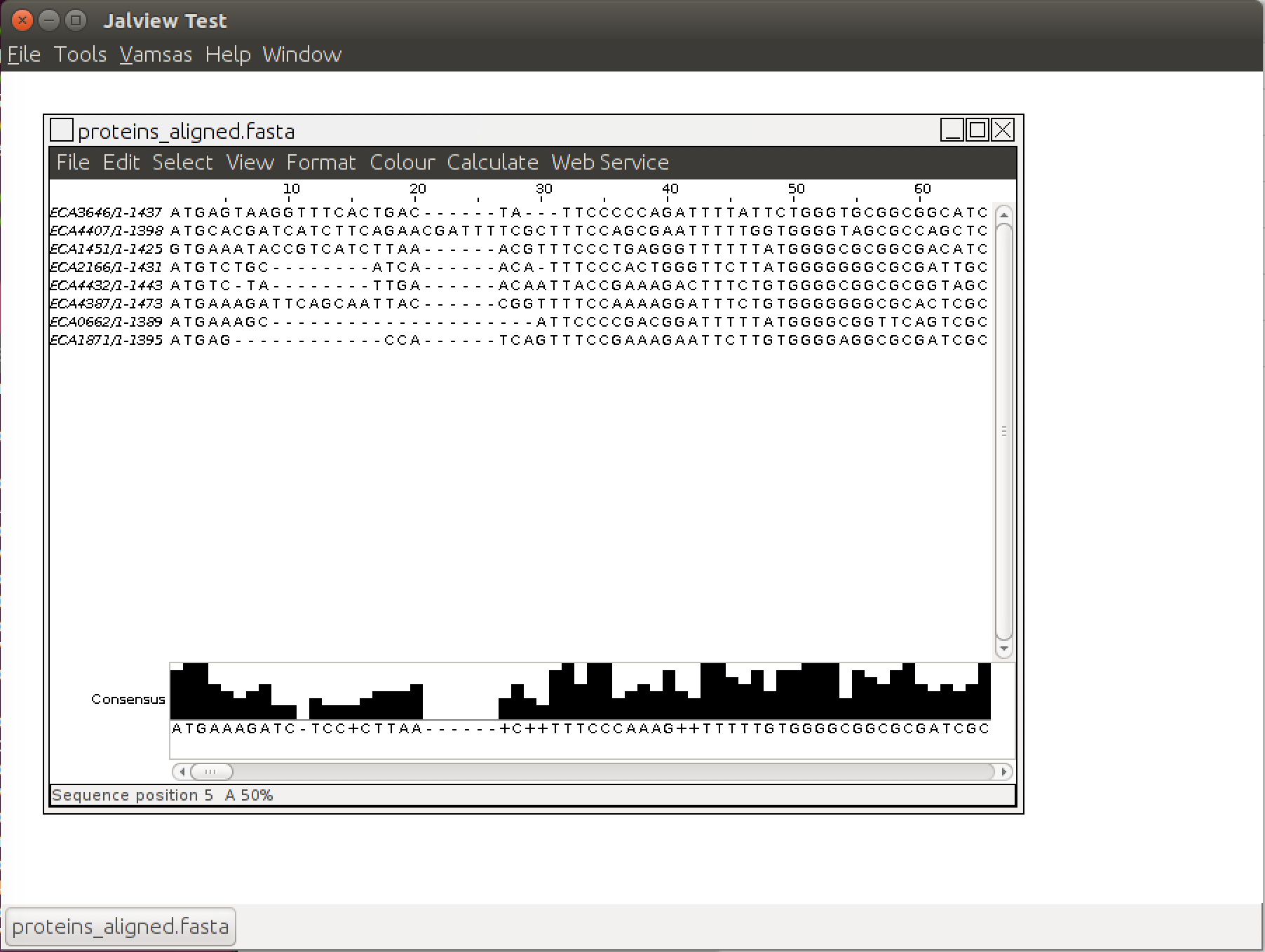
Try using the "Colour" menu to brighten this up and help show where these proteins agree with each other.